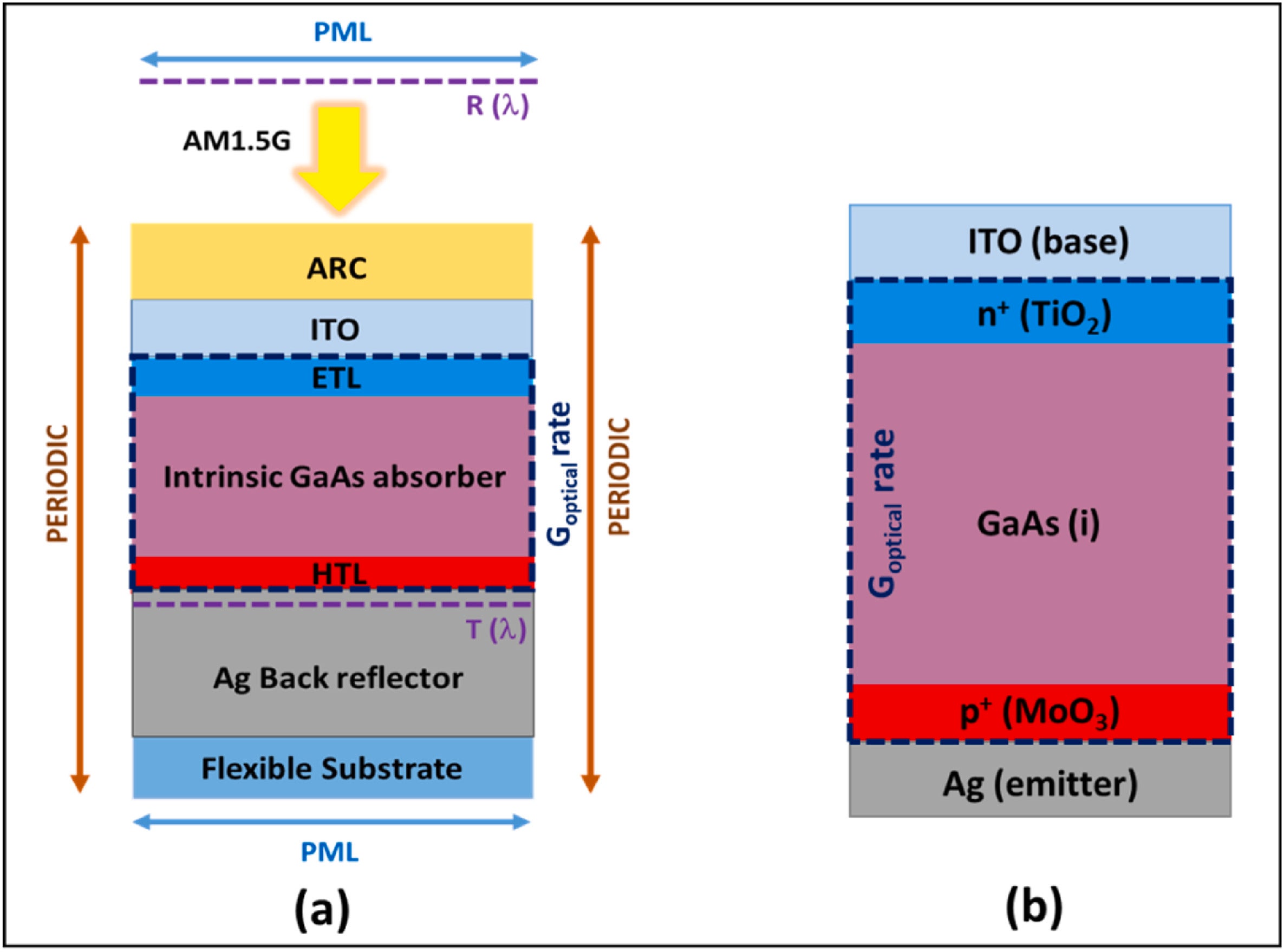
I am trying to design and simulate the above solar cell structure with FDTD and CHARGE module, with the following layers:
- TiO2 (10 nm): Electron transport layer (ETL), doped n-type (3e+19 cm−3).
- GaAs (290 nm): Absorber layer, lightly doped p-type (1e+16 cm−3).
- MoO3 (10 nm): Hole transport layer (HTL), doped p-type (1e+19 cm−3).
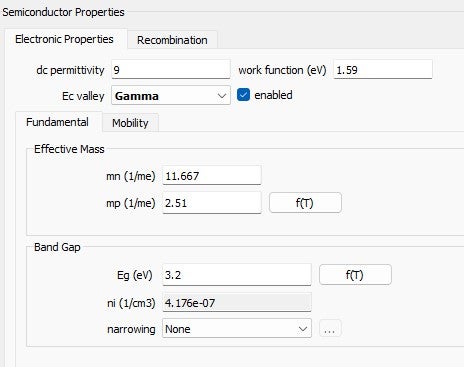
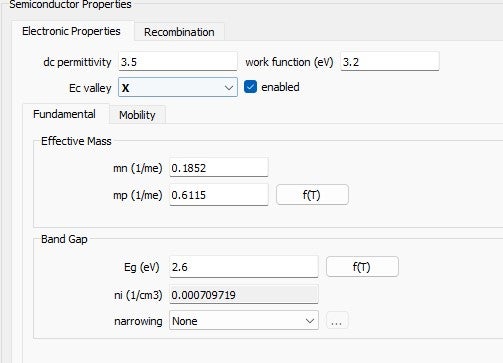
The semiconductor properties are defined as:: TiO₂: Data from Fig. 2.MoO₃: Data from Fig. 3.
I successfully simulated the structure in the FDTD module and generated the optical generation file (.mat) with mesh order = 2. However, while running the simulation in CHARGE, I encountered the following errors:
DESKTOP-URS3EQ3(process 0): The program terminated due to an error: Initialization failed to converge charge update. For instructions on how to troubleshoot this issue, please refer to this: online reference
DESKTOP-URS3EQ3(process 0): Error: there was an unknown parallel error. The error code is 9002, the process number is 0
I tried every possible combination from this link: https://optics.ansys.com/hc/en-us/articles/8355464963987-Troubleshooting-convergence-errors-in-CHARGE
Request for Assistance:
- Are there specific parameter adjustments (e.g., mobility models, recombination rates, or solver settings) recommended for this structure?
- Should I consider modifying the mesh or doping profiles for thin layers like TiO₂ and MoO₃?
- How can I resolve the initialization convergence error effectively?




















