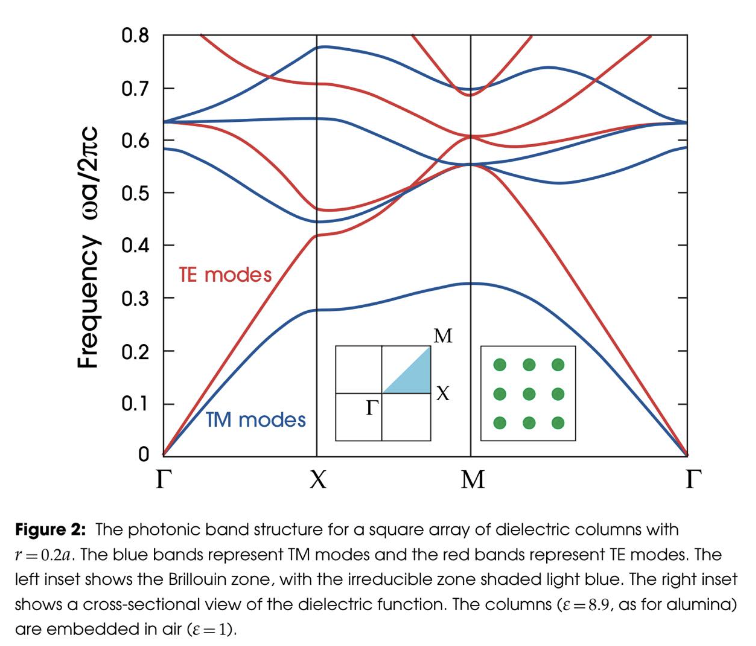
I have noticed a strong disagreement in bandstructure calculation in comparison to MIT photonic bands.
To demonstrate this, here is the bandstructure obtained from the band structure example: https://optics.ansys.com/hc/en-us/articles/360041566614-Rectangular-Photonic-Crystal-Bandstructure
Square lattice 2D TM mode:
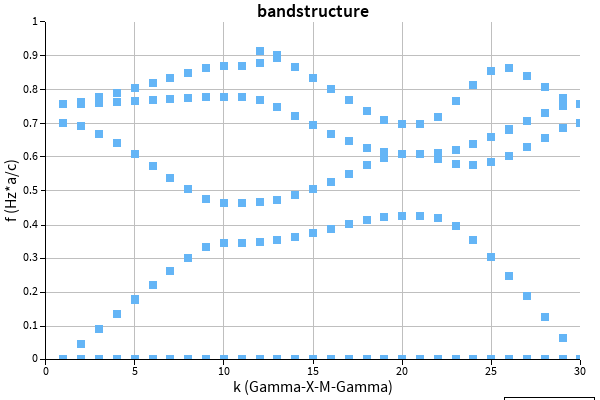
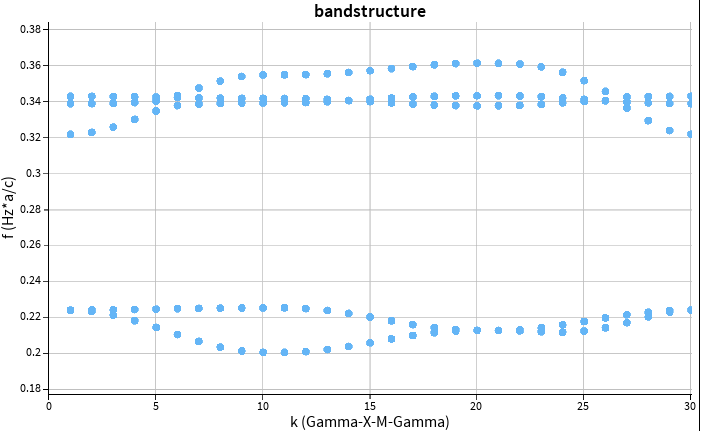
Calculating a bandstructure of exactly the same structure using MPB (https://mpb.readthedocs.io/en/latest/) I obtain: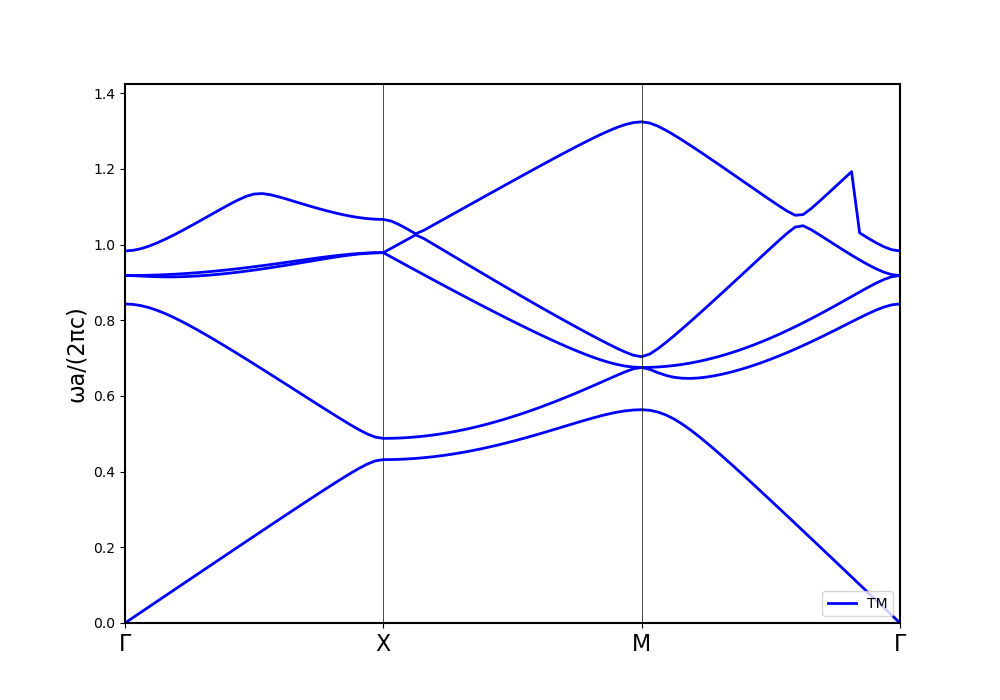
Those band structures are more or less in agreement, the shape is identical, and the energy axis seems to be a little scaled (apparent at 0.4 energy scale)
Now, increasing the refractive index in both solvers to 10 leads to the following results:
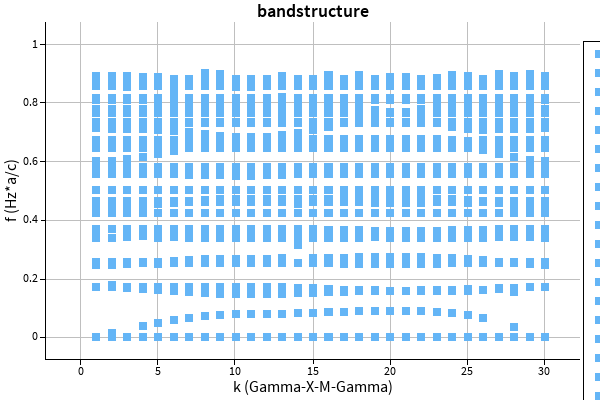
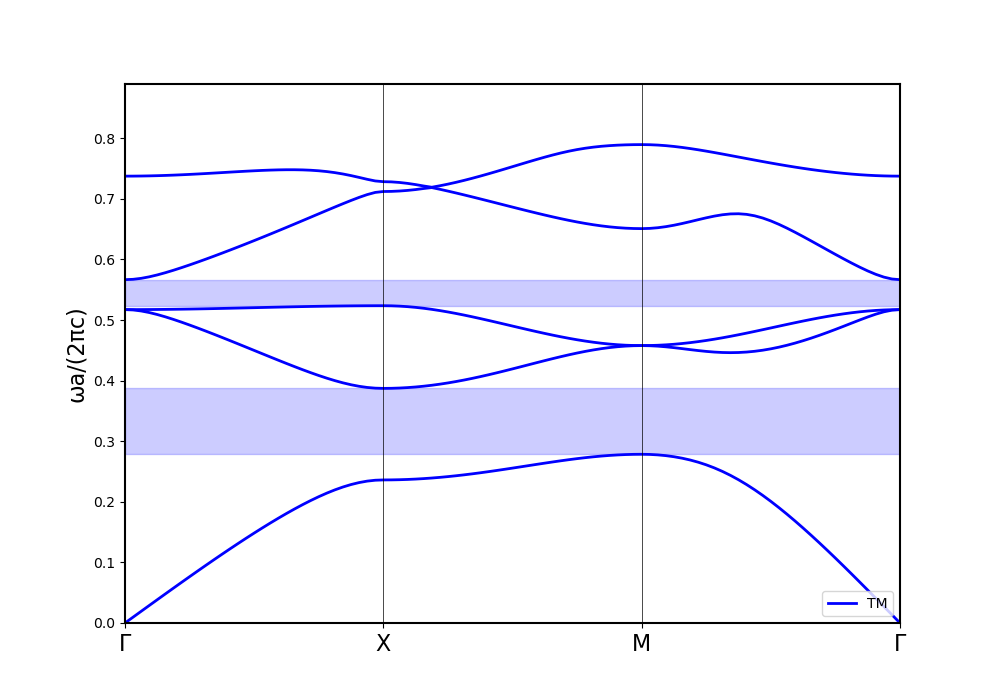
The shape of the bands is similar again, but this time, the energy axis is drastically scaled. Other calculation methods hint towards MPB being correct.
How do I combat this issue of energetical disagreement for high permittivity simulations?
Thank you,
Martin